![]()
There are now a number of molecular biology programs which may be of use to researchers at the institute. Some of these (eg VectorNTI) will be installed on your computer as part of the institute standard desktop. The rest are not installed by default and this document explains how you can access them.
Rather than having to install each package separately on everyones machine, the Bioinformatics group have set up a software repository on one of the site servers. Everyone can run these programs from that server, which means that the Bioinformatics group can take care of all the configuration and updating of these various programs.
If anyone has any suggestions for other packages which could be included in this repository, then we would be glad to hear then. Please contact a member of the bioinformatics group.
To install all of the packages on your machine you need to do the following:
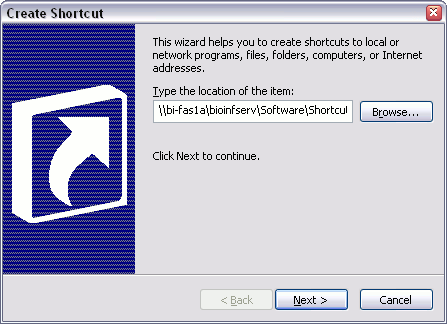
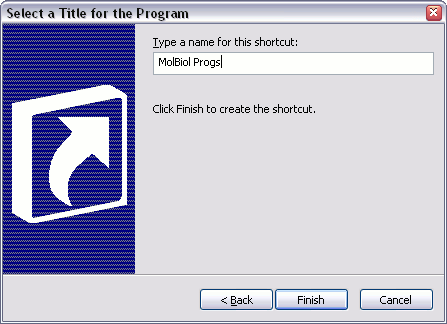
You should now have a folder on your desktop, inside which are all the icons to start the various molbio programs.
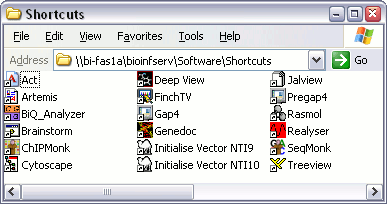
Artemis and ACT are tools developed by the Sanger Centre to help with the viewing and analysis of chromosome-sized chunks of DNA sequence.
Artemis is a genome browser, which allows you to view an annotated genome segment. It gives you control over the amount of detail you can see, and permits you to select gropus of features via a variety of tools. The program also allows you to perform your own analyses on the sequence via tools such as BLAST. You can also add your own annotation to the sequences.
ACT is a genome comparison tool which allows you to compare corresponding regions of two genomes via a synteny plot. Both of the genome segments are displayed in a cut down verision of the Artemis browser.
In order to compare sequencs you need to generate a comparion file. This only needs to be done once for each pair of sequences, and can be re-read whenever you want to look at them. Babraham have a web-based tool for generating this comparison file which can be found here.
If you're interested in learning more about ACT then you should consider registering your interest in our ACT training course.
This is a package which aids people involved in the analysis of bisulphite sequencing data, used to determine methylation patterns in DNA.
More information about BiQ analyser can be found here.
ChIPMonk is a program, written at Babraham, for the analysis of ChIP-on-chip experiment data. It provides a host of normalisation and analysis tools as well as a built in genome browser which allows you to view your data in the context of an annotated genome.
More information about ChIPMonk can be found here.
Cytoscape is a program which allows you to visualise data networks. It is most often used to view the output from programs which build gene regulation networks (often from microarray datasets).
More information about Cytoscape can be found here.
Deep View is a powerful molecular modelling package which allows you to visualise single and multiple protein structures in a variety of ways. In addition the program has the ability to calculate surfaces, electrostatics and to do in silico mutagenesis experiments. It also provides an interface to a homology modelling server to allow the prediction of structure for novel sequences.
In combination with the tools available in the programs section of the Bioinformatics intranet you can also use Deep View to generate publication quality molecular graphics, and even molecular animations.
If you're interested in learning more about Deep View then you should consider registering your interest in our Deep View training course.
More information about Deep View can be found here.
FinchTV is a simple sequencing trace viewer. You can use it by dropping a trace file onto the FinchTV icon to view it. FinchTV should only be used to get a quick view of your trace files. If you are using FinchTV to check for mutations in your sequence, then you really should be using the Staden package instead (see above).
More information about FinchTV can be found here.
Genedoc is a pretty versatile multiple sequence editor which runs on the desktop. Whilst it contains a great many features which you may find useful, it is primarily included because of its ability to read GCG format MSF files, and turn them into RSF files, which can be loaded directly into Word. It is therefore extremely useful for formatting multiple sequence alignments for inclusion into papers / theses etc.
If you're looking to export multiple alignments into reports using Genedoc then you should read the guide we have prepared on this subject
More information about Genedoc can be found here.
Jalview is a multiple alignment viewer and editor which can read pretty much all formats of multiple alignment. It has lots of options for shading your alignments according to different properties and can connect to various online databases to perform analysis on either the individual sequences or the alignment as a whole.
More information about Jalview can be found here.
Rasmol is a lightweight protein structure viewing program, which is a simpler alternative to the Deep View software mentioned above. It allows you to view protein structures in a number of different representations.
More information about Rasmol can be found here.
Realyser is a program to aid in the normalisation of QPCR data. It uses the data file from the ABI QPCR machine and helps the user select the best comination of controls from a panel of housekeeping genes. It then normalises your data using these controls, allowing you to proceed with your analysis.
SeqMonk is a program which can be used for the visualisation and analysis of mapped sequencing data. It is most often used to view the results of next-generation sequencing experiments.
More information about SeqMonk can be found here.
The Staden package is designed to help with sequence assembly problems. It is ideal for anyone taking on a sequencing project of any size, anything from confirming the sequence of a 500bp insert, to the resequencing of a 1Mb BAC. Staden works with either plain text sequence files, or chromatogram files from a sequencing service. It generates an assembly for you, and where present, will provide instant access to any undelying traces so you can see whether any mutations which show up are real or not.
If you're interested in learning more about Staden then you should consider registering your interest in our sequence assembly training course. More information about Staden can be found here.
Tree View is a program which allows the flexible construction of phylogenetic trees from the tree text files generated by packages such as Phylip. Trees generated by the program can be exported in common office applications, and are of publication quality.
More information about Tree View can be found here.
Xmenu is a small program which allows you to start the various software packages installed on the site Molecular Biology Unix server (Bihpc1). Double clicking on this icon will produce a prompt asking for your Bio4 username and password. Entering these correctly should produce the Xmenu window.
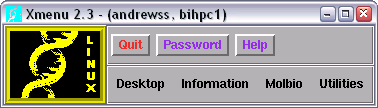
For more information about the Xmenu system please consult this separate guide.