Overview
HICUP - GENERAL INFORMATION
This documentation describes HiCUP, a bioinformatics pipeline produced by the Babraham Institute for processing Hi-C data. The documentation has three sections:
-
Overview - an explanation of Hi-C and how HiCUP helps analyse this kind of data
-
Quick Start - how to run HiCUP
-
Test Dataset - enables users to test HiCUP on their system
-
Scripts Description - details each step of the pipeline and how to run various HiCUP components on their own. Also discusses how to analyse Hi-C protocol variants.
To use HiCUP we suggest you read the Overview, the Quick Start and then follow the instructions to process the Test Dataset. The Scripts Description is usually only required for reference should you wish to understand an aspect of HiCUP in more detail.
There are also HiCUP tutorials on the Babraham Bioinformatics YouTube Channel. We recommend you watch these since watching instructional videos is often more clear than reading a manual:
-Tutorial 1: HiCUP Overview
-Tutorial 2: How to run HiCUP
-Tutorial 3: Interpreting HiCUP Summary Results
HI-C OVERVIEW
Chromosome folding can bring distant elements – such as promoters and enhancers – close together, which may affect genome activity. Consequently, investigating genomic conformation may improve understanding of processes such as transcription and replication.
Hi-C, developed from 3C, identifies long-range genomic interactions. The Hi-C protocol involves formaldehyde-fixing cells to create DNA-protein bonds that cross-link interacting DNA loci. The DNA is then digested with a restriction enzyme, fragmenting the molecule but maintaining the cross-links. The resulting overhanging 5’ ends of the DNA fragment are then filled-in with the concomitant incorporation of a biotinylated residue, followed by blunt-end ligation. This produces a library of ligation products that represents DNA restriction fragments that were close to each other in the nucleus at the moment of fixation. The library is then further fragmented, either by using a second restriction enzyme or, much more usually, by sonication. The Hi-C fragments are then pulled-down with streptavidin beads, which adhere with great affinity to the biotin tag at the ligation junction. The purified Hi-C fragments (termed di-tags) are then sequenced (Lieberman-Aiden et al.)
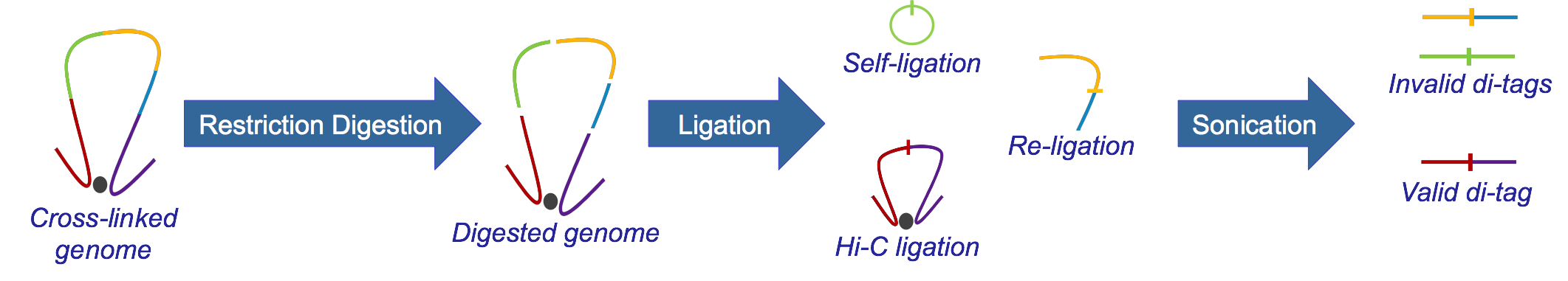
WHAT IS HICUP?
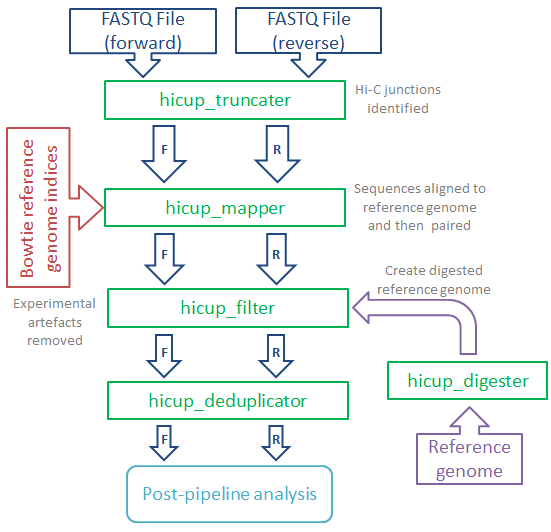
HiCUP is a bioinformatics pipeline for processing Hi-C data. The pipeline takes and maps FASTQ data against a reference genome and filters out frequently encountered experimental artefacts. The pipeline produces paired-read files in SAM/BAM format, each read pair corresponding to a putative Hi-C di-tag. HiCUP also produces summary statistics at each stage of the pipeline providing quality control, helping pinpoint potential problems and refine the experimental protocol.
The filtered read-pairs produced by HiCUP may then be used by Hi-C analysis tools to build a three-dimensional interpretation of the dataset. Such analysis tools include Hicpipe which eliminates pre-defined systematic biases to produce normalised contact maps; Homer, which also produces corrected contact maps without the need for specifying the global biases inherent to the Hi-C protocol. There is also a Bioconductor package named GOTHiC to identify statistically significant Hi-C interactions. Another useful tool is CHiCAGO, for identifying statistically significant interactions in Capture HiC (CHiC) data.
Another Babraham Institute project, SeqMonk, is an interactive genome browser that imports the SAM/BAM output from HiCUP.
HiCUP (Hi-C User Pipeline) comprises six Perl scripts for analysing Hi-C sequence data:
- HiCUP Digester - creates a digested reference genome
- HiCUP - executes sequentially the scripts below
- HiCUP Truncater - cuts reads at the putative Hi-C ligation junction
- HiCUP Mapper - aligns read pairs independently to a reference genome
- HiCUP Filter - removes commonly encountered Hi-C artefacts
- HiCUP Deduplicator - removes (retaining one copy) putative PCR duplicates
The pipeline enables multiplexed sequences to be sorted and mapped to the genome, the pairing of Hi-C fragment ends and filtering for valid Hi-C interaction products.
How to cite HiCUP
Wingett S, et al. (2015) HiCUP: pipeline for mapping and processing Hi-C data F1000Research, 4:1310 (doi: 10.12688/f1000research.7334.1)
Papers Citing HiCUP
Baxter JS, et al. (2018) Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 9(1), 1028, doi: 10.1038/s41467-018-03411-9
Novo CL, et al. (2018) Long-Range Enhancer Interactions Are Prevalent in Mouse Embryonic Stem Cells and Are Reorganized upon Pluripotent State Transition Cell Rep. 22(10), 2615-2627
Rennie S, et al. (2018) Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat Commun 9(1), 487
Burren OS, et al. (2017) Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol. 18(1), 165 doi: 10.1186/s13059-017-1285-0
Rubin AJ, et al. (2017) Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet. 49(10), 1522-1528
Petersen R, et al (2017) Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat Commun. doi:10.1038/ncomms16058
Nagano T, et al (2017) Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547(7661), 61-67
Siersbaek R, et al. (2017) Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol Cell.66(3):420-435
Freire-Pritchett P, et al. (2017) Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife doi:10.7554/eLife.21926
Moquin SA, et al (2017) The Epstein-Barr virus episome maneuvers between nuclear chromatin compartments during reactivation. Journal of Virology, doi: 10.1128/JVI.01413-17
Sud A, et al. (2017) Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nature Communications 8(1), 1892
Ke Y et al. (2017) 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 170(2):367-381
Litchfield K, et al. (2017) Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nature Genetics 49(7), 1133-1140
Gabriele M et al, (2017) YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. American Journal of Human Genetics 100(6), 907-925
El-Sharnouby S et al. (2017) Regions of very low H3K27me3 partition the Drosophila genome into topological domains. PloS one 12(3), e0172725
Law PJ, et al. (2017) Genome-wide association analysis of chronic lymphocytic leukaemia, Hodgkin lymphoma and multiple myeloma identifies pleiotropic risk loci. Scientific Reports 7(41071)
Aymard F, et al. (2017) Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology 24(4), 353-361
Javierre BM, et al. (2016) Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters Cell 167(5), 1369-1384
Cairns J, et al. (2016) CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 17(1), 127
Andrey G, et al. (2016) Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Research 27(2), 223-233
Franke M, et al. (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538(7624), 265-269
Krueger F, et al. (2016) SNPsplit: Allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Research 5:1479 DOI: 10.12688/f1000research.9037.2
Wu HJ, et al. (2016) A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 32(24), 3695-3701
Veluchamy A, et al. (2016) LHP1 Regulates H3K27me3 Spreading and Shapes the Three-Dimensional Conformation of the Arabidopsis Genome. PLoS One 11(7):e0158936
Schoenfelder S, et al. (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics, 47(10), 1179–1186
Mifsud B, et al. (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics, 47(6), 598-606
Nagano T, et al. (2015) Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology, 16(1), 175
Sahlén P, et al. (2015) Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology, 16(1), 156
Schoenfelder, S. et al. (2015) The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research, 25(4), 582-97
Chandra, T. et al. (2015) Global Reorganization of the Nuclear Landscape in Article Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Reports, 10(4), 1–13
Dryden, N. H. et al. (2014) Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research, 24(11), 1854-1868
We welcome your comments or suggestions, please email them to: steven.wingett@babraham.ac.uk