
HiCUP (Hi-C User Pipeline)
| Function | A tool for mapping and performing quality control on Hi-C data |
|---|---|
| Language | Perl/R |
| Requirements | Bowtie2 Perl R (with modules Tidyverse and Plotly installed) SAMtools Unix-based operating system gzip |
| Code Maturity | Beta - HiCUP is routinely being used to process real data, however it is still under active development. |
| Code Released | Yes, under GNU GPL v3 or later. |
| Initial Contact | Steven Wingett |
| Full Documentation | Full documentation and audio-visual demonstrations are available by clicking here |
Download Now |
|
Hi-C, developed from 3C, identifies long-range genomic interactions. The Hi-C protocol involves formaldehyde-fixing cells to create DNA-protein bonds that cross-link interacting DNA loci. The DNA is then digested and ligated to generate a library of products that were spatially close to each other in the nucleus.
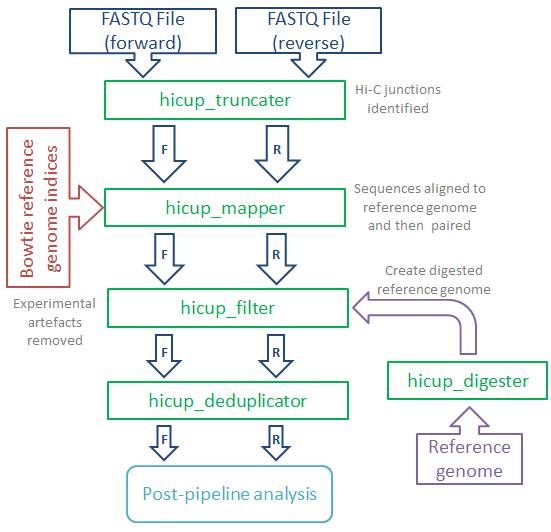
HiCUP is designed to take the raw sequence output from a HiC experiment and produce a filtered set of mapped interaction pairs, suitable for subsequent analysis. It will also produce a set of metrics which can be used to assess the quality of the data and help improve the construction of future libraries.
Main features:
- Identifies putative Hi-C junctions in sequence reads and truncates accordingly with a view to improving mapping efficiency
- Maps each read end independently using parameters suitable for Hi-C datasets
- Pairs forward and reverse reads for each di-tag, producing output
in SAM or BAM format
- Filters data to remove common artefacts e.g. di-tags where both reads map to the same restriction fragment
- Compatible with restriction enzyme/sonication or restriction enzyme-only protocols
Changelog
Attention: HiCUP 0.8.0 has major changes to the report summary generation. Systems now need R and the R modules Tidyverse and Plotly installed See documentation for further details- Click here for notes on all releases Version 0.8.0 onwards
- 18-06-20: Version 0.7.4 released
-
- Fixed bug when specifying a cut-site containing an N (any nucleotide). The script hicup_truncater used to incorporate such Ns into the FASTQ truncated read. This is now fixed and no longer occurs.
- HiCUP now uses CIGAR string information when positioning reads to restriction fragments and during the de-duplication process.
- Added scripts for GitHub Actions unit testing.
- Added a HiCUP Singularity recipe to the Misc folder.
- 13-05-20: Version 0.7.3 released
-
- Added hicup2juicer to make HiCUP output compatible with Juicer
- Modification to hicup_deduplicator output so files strictly adhere to SAM/BAM format
- Fixed bug preventing --nofill option from working
- No longer reports Bowtie2 message concerning gzbuffer changes
- 15-01-19: Version 0.7.2 released
-
- Fixed bug causing output files to not strictly adhere to SAM/BAM format.
- 12-11-18: Version 0.7.1 released
-
- Added the option --arima to HiCUP Digester, to generate digest files compatible with the Arima protocol.
- 25-10-18: Version 0.7.0 released
-
- HiCUP can now process restriction enzymes that cut at different sites. Such sequences contain an "N" within the restriction enzyme recognition site to denote the four bases (AGCT). Thanks to Rola Dali, Edouard Henrion and Mathieu Bourgey (McGill University for adding this feature.
- 10-05-18: Version 0.6.1 released
-
- Bowtie2 may now align reads using more than one thread (while preserving the input/output read order), thereby reducing processing times. The number of threads allocated to Bowtie2 will be the number of threads specified by the user divided by the number of files processed.
- Fixed bug causing HiCUP Mapper to try to read beyond the end of a mapped reads SAM file, consequently causing the script to crash.
- 23-04-18: Version 0.6.0 released
-
- Parameters adjusted for HiCUP Mapper in determining what constitutes a multi-mapping read, when using Bowtie2 as the aligner
- 26-10-17: Version 0.5.10 released
-
- Fixed bug causing hicup_mapper to not pair a small proportion of valid reads
- 15-09-16: Version 0.5.9 released
-
- Modified HiCUP Deduplicator so it may process genomes comprising hundreds of chromosomes (e.g. genome assemblies)
- Added script Misc/get_captured_reads for identifying on/off target di-tags in capture Hi-C datasets
- Fixed bug when running hicup_truncater independently, causing the script to interpret NoFill:0 in the config file as effectively NoFill:1
- 19-10-15: Version 0.5.8 released
-
- Fixed bug preventing an odd number of files being specified when using a configuration file with hicup_digester, hicup_filter or hicup_deduplicator.
- 18-06-15: Version 0.5.7 released
-
- Fixed bug preventing whole pipeline running to completion if --outdir is set to a folder other than the current working directory
- 09-06-15: Version 0.5.6 released
-
- Fixed bug preventing, when multiple samples are processed, the generation of HTML summary reports and the text file auditing the progress of read pairs through the entire pipeline
- 20-05-15: Version 0.5.5 released
-
- HiCUP produces a text file auditing the progress of read pairs through the entire pipeline for all samples processed
- Added the 'Conversion' folder, containing scripts for converting HiCUP BAM/SAM output into formats compatible with GOTHiC, Homer, Hicpipe and Fit-Hi-C
- Fixed bug preventing users specifying two --re1 restriction enzymes (i.e. the enzymes that create the Hi-C ligation junctions)
- 01-04-15: Version 0.5.4 released
-
- HiCUP Digester checks the first line of each input file begins with a valid FASTA header
- Fixed bug causing HiCUP to ignore choice of aligner
- HiCUP output compatible with SamTools release (v1.2), which is more strict with regard to the PG headers in a BAM/SAM file
- Fixed bug causing files to be compressed if zip:0 selected in a configuration file
- Fixed bug causing --re2 option sequence (i.e. not --re1 sequence) to be used to truncate reads prior to mapping
- Should one or more sample contain no valid di-tags the pipeline would produce no HTML summary files, even for samples containing valid di-tags. This no longer happens, instead the script displays a warning message reporting samples not progressing through the pipeline and produces HTML for samples containing valid di-tags
- Improved documentation by creating Markdown files. These were added to the 'Documentation' folder. The HiCUP Manual and QuickStart PDF files were deleted
- 31-12-14: Version 0.5.3 released
-
- HiCUP naming conventions changed to produce more succinct filenames
- 22-11-14: Version 0.5.2 released
-
- When running the whole pipeline, a folder may be specified to which all intermediate files are written. These files are deleted after the hicup_deduplicator script has terminated
- FASTQ format now specified differently, valid options are: Sanger, Solexa_Illumina_1.0, Illumina_1.3 or Illumina_1.5
- All configuration files kept in the 'config_files' folder
- hicup_digester may be passed lowercase characters denoting the restriction enzyme recognition site
- Pipeline no longer terminates if SAM Tools in not installed
- 02-11-14: Version 0.5.1 released
-
- hicup_deduplicator now accepts --threads as an argument
- 04-10-14: Version 0.5.0 released
-
- Compatible with Bowtie2
- To reduce mis-mapping, HiCUP no longer uses '--strata' as a parameter when aligning with Bowtie
- R used to generate figures instead of Perl module GD::Graph
- File pairs should now be placed on adjacent lines in their respective configuration files
- Command line arguments can be used in conjunction with a configuration file
- Fixed bug causing HiCUP only being able to process files in the current working directory
- 27-11-13: Version 0.4.2 released
-
- Pipeline produces di-tag length (prior to filtering) distribution plots in the HTML document and as a separate GD::Graph line plot
- Pipeline produces an HTML report when following the double-digest protocol
- HiCUP version number and pipeline parameters are printed in the headers of the SAM/BAM files
- User can specify hi-c ligation sequences directly via hicup.conf
- 20-09-13: Version 0.4.1 released
-
- Fixed bug causing results from different datasets to become mixed in the HTML report
- Improved HTML report layout
- 10-09-13: Version 0.4.0 released
-
- Added --nofill option to hicup_truncater (and consequently hicup) allowing users to specify that a fill-in of sticky ends was not performed
- hicup_filter now discriminates between same-fragment dangling ends and same-fragment internal
- Users can now specify an output directory to which output files are written
- HiCUP sonication protocol now generates an HTML report summarising the results of each file processed
- 02-11-12: Version 0.3.0 released
-
- hicup_sorter removed from the pipeline
- The pipeline determines automatically the FASTQ format adopted if not specified
- The pipeline determines the path to Bowtie if not specified by the user. Also, fixed a bug affecting how HiCUP identifies the location of SAMtools
- Improved how the pipeline checks Bowtie indices have been specified correctly
- 03-08-12: Version 0.2.2 released
-
- The mapping process is now less memory intensive
- HiCUP can process files in a separate folder from the hicup.conf configuration file
- The hicup master script terminates immediately if another pipeline script dies
- 19-07-12: Version 0.2.1 released
-
- hicup_filter reports the number of read-pairs generated by circularized restriction fragments
- hicup_filter reports the absolute number of read-pairs by category
- 26-06-12: Version 0.2.0 released
-
- Added new script 'hicup_deduplicator' for removing duplicate di-tags
- hicup_mapper and hicup_pairer combined into a single script
- hicup_filter, when processing Hi-C data generated using the Hi-C sonication protocol, now rejects di-tags on the basis of size AFTER all other filters have been passed
- hicup_filter and hicup_deduplicator produce pie charts summarising the results
- hicup_filter modified so when following the sonication protocol it identifies and rejects di-tags containing re-ligated fragments, not simply those on adjacent fragments
- hicup_truncater now reports the average length of truncated sequences
- Fixed a bug causing hicup_digester to only process the last chromosome in a file containing multiple chromosomes
- 22-03-12: Version 0.1.1 released
-
- Initial release
- All basic functions working