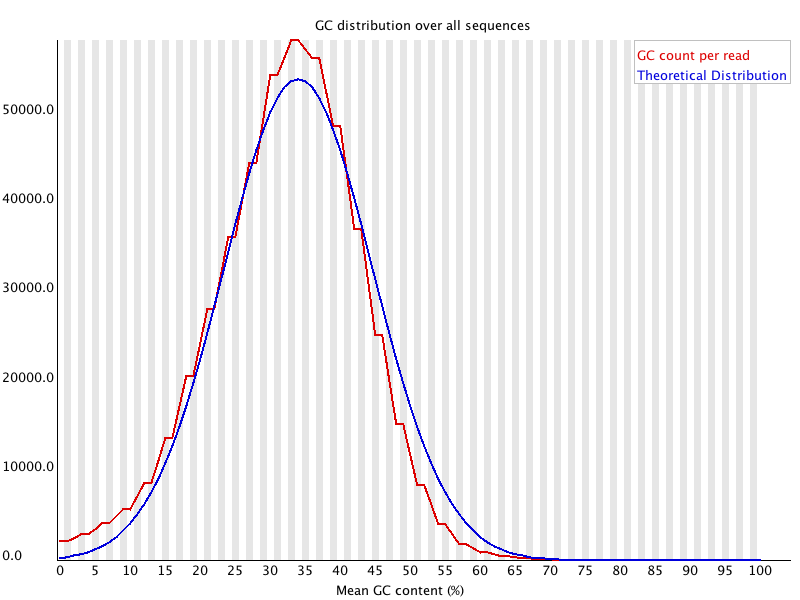
This module measures the GC content across the whole length of each sequence in a file and compares it to a modelled normal distribution of GC content.
In a normal random library you would expect to see a roughly normal distribution of GC content where the central peak corresponds to the overall GC content of the underlying genome. Since we don't know the the GC content of the genome the modal GC content is calculated from the observed data and used to build a reference distribution.
An unusually shaped distribution could indicate a contaminated library or some other kinds of biased subset. A normal distribution which is shifted indicates some systematic bias which is independent of base position. If there is a systematic bias which creates a shifted normal distribution then this won't be flagged as an error by the module since it doesn't know what your genome's GC content should be.
A warning is raised if the sum of the deviations from the normal distribution represents more than 15% of the reads.
This module will indicate a failure if the sum of the deviations from the normal distribution represents more than 30% of the reads.
Warnings in this module usually indicate a problem with the library. Sharp peaks on an otherwise smooth distribution are normally the result of a specific contaminant (adapter dimers for example), which may well be picked up by the overrepresented sequences module. Broader peaks may represent contamination with a different species.