HiCUP Overview¶
ATTENTION: HiCUP Version 8+ generates new format summary report HTML and text files. You will also notice new a pipeline script HiCUP Reporter and a new R markdown file r_scripts/hicup_reporter.rmd. To generate the HTML summary report now requires R to be installed on your system as well as the R modules Tidyverse and Plotly. Also, collated summary reports will no longer be generated for the rarely used for double-digest Hi-C protocol.
General Information¶
This documentation describes HiCUP, a bioinformatics pipeline produced by the Babraham Institute for processing Hi-C data. The documentation has three sections:
- Overview - an explanation of Hi-C and how HiCUP helps analyse this kind of data
- Quick Start - how to run HiCUP
- Test Dataset - enables users to test HiCUP on their system
- Scripts Description - details each step of the pipeline and how to run various HiCUP components on their own. Also discusses how to analyse Hi-C protocol variants.
To use HiCUP we suggest you read the Overview, the Quick Start and then follow the instructions to process the Test Dataset. The Scripts Description is usually only required for reference should you wish to understand an aspect of HiCUP in more detail.
There are also HiCUP tutorials on the Babraham Bioinformatics YouTube Channel. We recommend you watch these since watching instructional videos is often more clear than reading a manual:
Tutorial 3: Interpreting HiCUP Summary Results
We welcome your comments or suggestions, please email them to: steven.wingett@babraham.ac.uk
Hi-C Overview¶
Chromosome folding can bring distant elements – such as promoters and enhancers – close together, which may affect genome activity. Consequently, investigating genomic conformation may improve understanding of processes such as transcription and replication.
Hi-C, developed from 3C, identifies long-range genomic interactions. The Hi-C protocol involves formaldehyde-fixing cells to create DNA-protein bonds that cross-link interacting DNA loci. The DNA is then digested with a restriction enzyme, fragmenting the molecule but maintaining the cross-links. The resulting overhanging 5’ ends of the DNA fragment are then filled-in with the concomitant incorporation of a biotinylated residue, followed by blunt-end ligation. This produces a library of ligation products that represents DNA restriction fragments that were close to each other in the nucleus at the moment of fixation. The library is then further fragmented, either by using a second restriction enzyme or, much more usually, by sonication. The Hi-C fragments are then pulled-down with streptavidin beads, which adhere with great affinity to the biotin tag at the ligation junction. The purified Hi-C fragments (termed di-tags) are then sequenced (Lieberman-Aiden et al.)
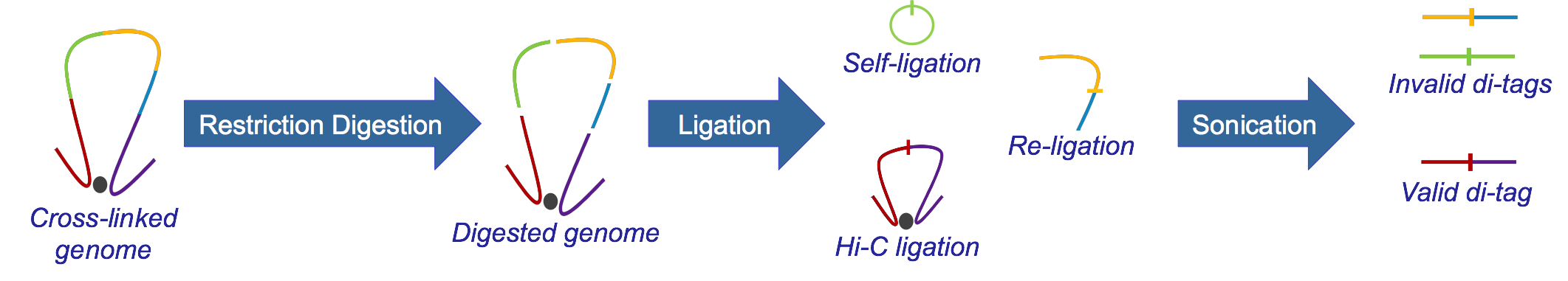
What is HiCUP?¶
HiCUP is a bioinformatics pipeline for processing Hi-C data. The pipeline takes and maps FASTQ data against a reference genome and filters out frequently encountered experimental artefacts. The pipeline produces paired-read files in SAM/BAM format, each read pair corresponding to a putative Hi-C di-tag. HiCUP also produces summary statistics at each stage of the pipeline providing quality control, helping pinpoint potential problems and refine the experimental protocol.
The filtered read-pairs produced by HiCUP may then be used by Hi-C analysis tools to build a three-dimensional interpretation of the dataset. Such analysis tools include Hicpipe which eliminates pre-defined systematic biases to produce normalised contact maps; Homer, which also produces corrected contact maps without the need for specifying the global biases inherent to the Hi-C protocol. There is also a Bioconductor package named GOTHiC to identify statistically significant Hi-C interactions. Another useful tool is CHiCAGO, for identifying statistically significant interactions in Capture HiC (CHiC) data.
Another Babraham Institute project, SeqMonk, is an interactive genome browser that imports the SAM/BAM output from HiCUP.
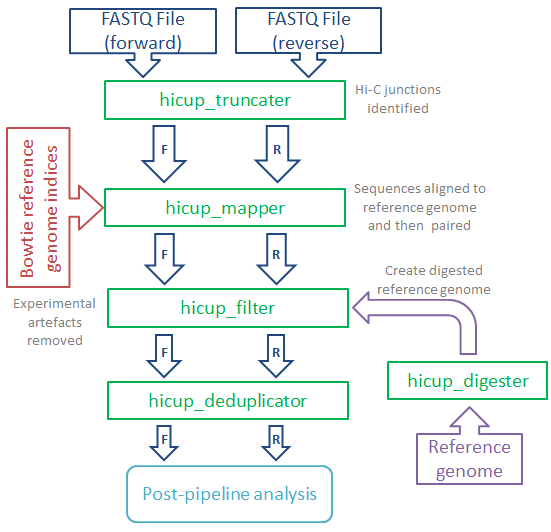
HiCUP (Hi-C User Pipeline) comprises six Perl scripts for analysing Hi-C sequence data:
- HiCUP Digester - creates a digested reference genome
- HiCUP - executes sequentially the scripts below
- HiCUP Truncater - cuts reads at the putative Hi-C ligation junction
- HiCUP Mapper - aligns read pairs independently to a reference genome
- HiCUP Filter - removes commonly encountered Hi-C artefacts
- HiCUP Deduplicator - removes (retaining one copy) putative PCR duplicates
The pipeline enables multiplexed sequences to be sorted and mapped to the genome, the pairing of Hi-C fragment ends and filtering for valid Hi-C interaction products.
HiCUP Quick Start Guide¶
HiCUP is a bioinformatics pipeline for processing Hi-C data. The pipeline maps FASTQ data against a reference genome and filters out frequently encountered experimental artefacts. The pipeline produces paired-read files in SAM/BAM format, each read pair corresponding to a putative Hi-C di-tag. HiCUP also produces summary statistics at each stage of the pipeline providing quality control, helping pinpoint potential problems and refine the experimental protocol.
Requirements¶
HiCUP should work on most Linux-based operating systems. It requires a working version of Perl and uses Bowtie or Bowtie2 to perform the mapping.
Full functionality requires R (tested with version 3.1.2) and SAM tools (version 0.1.18 or later).
Memory requirements depend on the size of the input files, but as a rough guide allocating 2Gb of RAM per file processed simultaneously (defined by –threads argument) should suffice.
Installation¶
HiCUP is written in Perl and executed from the command line. To install HiCUP download the hicup_v0.X.Y.tar.gz file and extract all files by typing:
tar -xvzf hicup_v0.X.Y.tar.gz
Check after extracting that the Perl scripts are executable by all, which can be achieved with the command:
chmod a+x [files]
Running HiCUP¶
1) Create Aligner Indices HiCUP uses the aligner Bowtie or Bowtie2 to map sequences to a reference genome, requiring the construction of genome index files. These indices MUST be constructed from the same reference genome files as used by the HiCUP Digester script.
On the command line enter ‘bowtie-build’ (or bowtie2-build) to construct the indices, followed by a comma-separated list of the sequence files and then a space followed by the name of the output indices:
bowtie-build 1.fa,2.fa,...,MT.fa Human_GRCh37
bowtie2-build 1.fa,2.fa,...,MT.fa Human_GRCh37
Refer to the Bowtie or Bowtie2 manuals for further guidance.
2) Create a digested reference genome To filter out common experimental artefacts, HiCUP requires the positions at which the restriction enzyme(s) used in the protocol cut the genome. The script HiCUP Digester creates this reference genome digest file. The example below performs an in silico HindIII digest of all DNA sequences contained within the files in the current working directory suffixed with ‘.fa’. The digest output file will be labelled as the genome ‘Human_GRCh37’. Provide the full path to HiCUP Digester or the sequence files to be digested if they are not in the current working directory.
Execute the script:
hicup_digester --genome Human_GRCh37 --re1 A^AGCTT,HindIII *.fa
The argument ‘–re1’ specifies the restriction enzyme used to digest the genome (a caret symbol ‘^’ is used to denote the restriction enzyme cut site, and a comma separates the DNA sequence from the restriction enzyme name). The argument ‘–genome’ is for specifying the name of the genome to be digested, it is NOT used to specify the path to the genome aligner indices.
Hi-C Protocol Variations: some Hi-C protocols may use two restriction enzymes at this stage (i.e. the creation of the initial Hi-C interaction). To specify two enzymes use the nomenclature: –re1 A^GATCT,BglII:A^AGCTT,HindIII
- Run the HiCUP Pipeline
Create an example HiCUP configuration file in your current working directory:
hicup --example
Use a text editor to edit the configuration file as required, such as in the following example:
#Directory to which output files should be
#written (optional parameter)
#Set to current working directory by default
Outdir:
#Number of threads to use
Threads: 1
#Suppress progress updates (0: off, 1: on)
#Quiet:0
#Retain intermediate pipeline files (0: off, 1: on)
Keep:0
#Compress outputfiles (0: off, 1: on)
Zip:1
#Path to the alignment program (Bowtie or Bowtie2)
#Remember to include the executable Bowtie/Bowtie2 filename.
#Note: ensure you specify the correct aligner i.e.
#Bowtie when using Bowtie indices, or Bowtie2 when using Bowtie2 indices.
#In the example below Bowtie2 is specified.
Bowtie2: /usr/local/bowtie2/bowtie2
#Path to the reference genome indices
#Remember to include the basename of the genome indices
Index: /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37
#Path to the genome digest file produced by hicup_digester
Digest: Digest_Mouse_genome_HindIII_None_12-32-06_17-02-2012.txt
#FASTQ format (valid formats: 'Sanger', 'Solexa_Illumina_1.0',
#'Illumina_1.3' or 'Illumina_1.5'). If not specified, HiCUP will
#try to determine the format automatically by analysing one of
#the FASTQ files. All input FASTQ will assumed to be in that
#format.
Format: Sanger
#Maximum di-tag length (optional parameter)
Longest: 700
#Minimum di-tag length (optional parameter)
Shortest: 50
#FASTQ files to be analysed, placing paired files on adjacent lines
s_1_1_sequence.txt
s_1_2_sequence.txt
s_2_1_sequence.txt
s_2_2_sequence.txt
s_3_1_sequence.txt.gz
s_3_2_sequence.txt.gz
Rename the configuration file if desired.
Enter the following text in the command line to run the whole HiCUP pipeline using the parameters specified in the configuration file:
hicup --config [Configuration Filename]
The –config flag is used to specify the configuration filename. Also, remember to provide the full path to the HiCUP script and/or the configuration file if they are not in the current working directory.
Please Note: HiCUP attempts to intelligently name files as the pipeline proceeds, so avoid passing HiCUP input files with identical names prior to the filename extension. For example, the files ‘sample.fa’ and ‘sample.fastq’ would produce files with identical names as the pipeline progresses. This problem could be overcome by renaming one the files to ‘sample2.fa’. To minimise inconvenience, HiCUP will immediately produce a warning message and not run if the input filenames are too similar.
- Output
The pipeline produces paired-read BAM files representing the filtered di-tags. HiCUP also generates an HTML summary report for each sample and a text file summarising every sample processed. Summary text files and SVG-format charts are also created at each step along the pipeline.
The ‘Conversion’ folder within the main HiCUP directory contains Perl scripts to convert HiCUP BAM/SAM files into a format compatible with other analysis tools. Executing one of these files with the command line argument –help prints instructions on how to use the conversion script.
Scripts Description¶
The rest of this section discusses each script in more detail and provides instructions on how to execute pipeline scripts separately i.e. without running the HiCUP control script.
The HiCUP pipeline comprises the following scripts:
- HiCUP (control script)
- HiCUP Truncater
- HiCUP Mapper
- HiCUP Filter
- HiCUP Deduplicator
- HiCUP Digester
HiCUP (pipeline control script)¶
The hicup Perl script controls the other programs in the HiCUP pipeline
Synopsis¶
hicup [OPTIONS]... [Configuration FILE]...
Function¶
The HiCUP pipeline comprises the scripts ‘hicup_truncater’, ‘hicup_mapper’, ‘hicup_filter’ and ‘hicup_deduplicator’ (‘hicup_digester generates the genome_digest file used by hicup_filter). The pipeline takes FASTQ files and generates Hi-C di-tag paired reads, aligned to a specified reference genome. The HiCUP script regulates the pipeline, executing each script in turn and passing output from one stage of the program to the next.
The designated configuration file sets the parameters for the whole pipeline. The configuration file lists the names of the FASTQ file pairs to be processed.
Configuration File Example:
#Directory to which output files should be written (optional parameter)
#Set to current working directory by default
Outdir:
#Number of threads to use
Threads: 1
#Suppress progress updates (0: off, 1: on)
Quiet:0
#Retain intermediate pipeline files (0: off, 1: on)
Keep:0
#Compress outputfiles (0: off, 1: on)
Zip:1
#Path to the alignment program (Bowtie or Bowtie2)
#Remember to include the executable Bowtie/Bowtie2 filename.
#Note: ensure you specify the correct aligner i.e.
#Bowtie when using Bowtie indices, or Bowtie2 when using Bowtie2 indices.
#In the example below Bowtie2 is specified.
Bowtie2: /usr/local/bowtie2/bowtie2
#Path to the genome digest file produced by hicup_digester
Digest: Digest_Mouse_genome_HindIII_None_12-32-06_17-02-2012.txt
#FASTQ format (valid formats: 'Sanger', 'Solexa_Illumina_1.0', 'Illumina_1.3' or 'Illumina_1.5')
#If not specified, HiCUP will try to determine the format automatically by analysing
#one of the FASTQ files. All input FASTQ will assumed to be in this format
Format: Sanger
#Maximum di-tag length (optional parameter)
Longest: 700
#Minimum di-tag length (optional parameter)
Shortest: 50
#FASTQ files to be analysed, placing paired files on adjacent lines
s_1_1_sequence.txt
s_1_2_sequence.txt
s_2_1_sequence.txt
s_2_2_sequence.txt
s_3_1_sequence.txt.gz
s_3_2_sequence.txt.gz
Command Line Example:
hicup --zip --bowtie /usr/local/bowtie/bowtie --index /data/Genomes/mm9/Mus_musculus.NCBIM37 --digest Digest_mm9_HindIII_None_12-32-06_17-02-2012.txt --format Sanger --longest 800 --shortest 150 s_1_1_sequence.txt s_1_2_sequence.txt s_2_1_sequence.txt s_2_2_sequence.txt s_3_1_sequence.txt.gz s_3_2_sequence.txt.gz``
This configuration instructs the pipeline to process and pair the files s_1_1_sequence.txt with s_1_2_sequence.txt; and s_2_1_sequence.txt with s_2_2_sequence.txt; and s_3_1_sequence.txt with s_3_2_sequence.txt. Remember, a file pair generates one output file.)
HiCUP also requires the paths to Bowtie and the genome digest file.
Command Line Options¶
--bowtie Specify the path to Bowtie --bowtie2 Specify the path to Bowtie 2 --config Specify the configuration file --digest Specify the digest file listing restriction fragment co-ordinates --example Produce an example configuration file --format Specify FASTQ format Options: Sanger, Solexa_Illumina_1.0, Illumina_1.3, Illumina_1.5 --help Print help message and exit --index Path to the relevant reference genome Bowtie/Bowtie2 indices --keep Keep intermediate pipeline files --longest Maximum allowable insert size (bps) --nofill Hi-C protocol did NOT include a fill-in of sticky ends prior to ligation step and therefore FASTQ reads shall be truncated at the Hi-C restriction enzyme cut site (if present) sequence is encountered --outdir Directory to write output files --quiet Suppress progress reports (except warnings) --shortest Minimum allowable insert size (bps) --temp Write intermediate files (i.e. all except summaryfiles and files generated by HiCUP Deduplicator) to a specified directory --threads Specify the number of threads, allowing simultaneous processing of multiple files --version Print the program version and exit --zip Compress output
HiCUP Truncater¶
The HiCUP Truncater Perl script terminates sequence reads at specified Hi-C ligation junctions
Synopsis¶
hicup_truncater [OPTIONS]... --config [Configuration FILE]...
hicup_truncater [OPTIONS]... [FASTQ FILES]...
Function¶
Valid Hi-C pairs comprise two DNA fragments from different regions of the genome ligated together. Typically, a forward read maps to one ligation fragment; the reverse read maps to the other. However, this is not always true since the Hi-C ligation junction may be found within the sequenced region. Such reads will most likely be removed from the Hi-C pipeline during the mapping process, thereby losing potentially valid data. The hicup_truncater script helps remedy this by identifying ligation junctions within reads and deleting sequence downstream of the restriction enzyme recognition site.

The names of the files to be processed and the restriction site may be passed to the scrip using a configuration file or command line arguments. The configuration file contains: i) the recognition sequence of the first (or only) restriction enzyme used in the Hi-C protocol and ii) the sequence files to be processed by the HiCUP Truncater.
Configuration File Example:
re1: A^GATCT s_6_1_sequence.txt_CCTT.fastq.gz s_6_2_sequence.txt_CCTT.fastq.gz
Command Line Example:
hicup_truncater --re1 A^GATCT,BglII s_6_1_sequence.txt_CCTT.fastq.gz s_6_2_sequence.txt_CCTT.fastq.gz
(The caret symbol (‘^’) denotes the cut position in the restriction enzyme recognition sequence.)
This configuration instructs the script to process the files ‘s_6_1_sequence.txt_CCTT.txt’ and ‘s_6_2_sequence.txt_CCTT.gz’. The script identifies any reads containing the Hi-C ligation sequence ‘AGATCGATCT’ (this sequence is not found in the original genomic sequence but is generated by restriction digestion with BglII, removal of sticky ends followed by blunt-ended ligation) and discards sequence downstream of the restriction cut site.
The program creates sequence files named the same as the input files, only suffixed with ‘trunc.fastq’. The script also produces a date-stamped summary file (e.g. ‘hicup_truncater_summary_08-59-17_30-01-2015.txt’) listing the number of reads truncated or not truncated for each input sequence file, along with accompanying SVG format charts.
Command Line Options¶
--config Name of the optional configuration file --help Print program help and exit --nofill Hi-C protocol did NOT include a fill-in of sticky ends prior to re-ligation and therefore reads shall be truncated at the restriction site sequence --outdir Directory to write output files --quiet Suppress all progress reports, except warnings --re1 Restriction enzyme recognition sequence --sequences Instead of specifying a restriction enzyme recognition sequence, specify the ligation sequences directly --threads Number of threads to use, allowing simultaneous processing of different files --version Print the program version and exit --zip Compress output
HiCUP Mapper¶
The HiCUP Mapper script aligns paired reads independently to a reference genome and retains reads where both partners align
Synopsis¶
hicup_mapper [OPTIONS]... --config [Configuration FILE]...
hicup_mapper [OPTIONS]... [FASTQ FILES]...
Function¶
Valid Hi-C ligation products comprise two restriction fragments from different regions of the genome ligated together. This program maps Hi-C di-tags against a reference genome to determine from where each restriction fragment is derived. Following mapping the forward and reverse reads are paired i.e. two input files result in one output file.
HiCUP Mapper uses the sequence alignment programs Bowtie or Bowtie2 to perform the mapping.
Bowtie mapping parameters:
-p 1 -n 1 -m 1 –best
-p 1: launches Bowtie with only one search thread. Although limiting the search to one processor is slower on multi-core machines, it ensures the order of the returned mapped reads is the same as found in the input file (ignoring omitted unmapped sequences), essential for the correct functioning of HiCUP. (Bowtie actually defaults to -p 1, but this has been made explicit in the Perl script due to the importance of preserving the read order.)
-n 1: alignments may have no more than 1 mismatch in the first 28 bases (seed) and the sum of the Phred quality values at all mismatched positions (not just in the seed) may not exceed 70. Bowtie rounds quality values to the nearest 10, saturating at 30.
-m 1: report only unique alignments
–best: reports alignments in best-to-worst order
The configuration file sets the parameters for HiCUP Mapper, it contains: i) names of files to be mapped; ii) local path to Bowtie; iii) path to the relevant reference genome Bowtie indices; iv) the sequence format.
Bowtie2 Mapping Parameters:
–very-sensitive: slower but a more sensitive and more accurate option
–no-unal: suppress SAM records for reads that failed to align.
–threads: number of threads specified by the user divided by the number of files processed
–reorder: ensure the read output order is the same as the input order when multi-threading
Bowtie2 does not have a direct equivalent of the -m 1 option available in the original Bowtie. Therefore, to identify and filter out multi-mapping reads, HiCUP processes the SAM file generated by Bowtie2. A reads is considered as uniquely mapping if the quality score is greater than or equal to 30 and either i) the read cannot be mapped to another location or ii) if the read can be mapped to other locations, then the difference in quality score between this hit and the next-best match should be at least 10 (as reported in the Bowtie2 SAM tags “AS” and “XS”). Before HiCUP version 0.6.1, a unique-mapping read was defined simply as having no next-best hit when using Bowtie2 as the aligner.
Configuration file example:
Bowtie: /usr/local/bowtie/bowtie
Index: /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37
Format: Sanger
Threads: 4
s_1_1_sequence.txt_CCTT_trunc
s_1_2_sequence.txt_CCTT_trunc
s_1_1_sequence.txt_AAGT_trunc
s_1_2_sequence.txt_AAGT_trunc
Command line example
hicup_mapper --bowtie /usr/local/bowtie/bowtie --index /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37 --format Sanger --threads 4 s_1_1_sequence.txt_CCTT_trunc s_1_2_sequence.txt_CCTT_trunc s_1_1_sequence.txt_AAGT_trunc s_1_2_sequence.txt_AAGT_trunc
The above example sends four files to Bowtie for mapping against the mouse NCBIM37 genome. Any text not preceded with a flag is assumed to be a sequence filename.
The FASTQ format options are:
- Sanger
- Solexa_Illumina_1.0
- Illumina_1.3
- Illumina_1.5
The output filenames will be based on the input filenames but suffixed with ‘.pair.sam’ or ‘.pair.bam’. The script also produces a date-stamped file (e.g. ‘hicup_mapper_summary_08-59-17_30-01-2015.txt’) and summarises the results in SVG graphical format.
Command Line Options¶
--bowtie Specify the path to Bowtie --bowtie2 Specify the path to Bowtie 2 --config Specify the configuration file --format Specify FASTQ format Options: Sanger, Solexa_Illumina_1.0, Illumina_1.3, Illumina_1.5 --help Print help message and exit --index Path to the relevant reference genome Bowtie/Bowtie2 indices --outdir Directory to write output files --quiet Suppress progress reports (except warnings) --threads Specify the number of threads, allowing simultaneous processing of different files (default: 1) --version Print the program version and exit --zip Compress output
HiCUP Filter¶
The HiCUP Filter Perl script classifies read pairs, identifying valid Hi-C di-tags
Synopsis¶
hicup_filter [OPTIONS] --config [Configuration FILE]...
hicup_filter [OPTIONS] [hicup_mapper output FILE]...
Function¶
The majority of reads generated by the HiCUP Mapper script are most likely valid Hi-C products, but a substantial minority are probably not and should be removed. The HiCUP Filter script processes paired reads together with the file created by HiCUP Digester to identify valid Hi-C pairs.
The names of the files to be processed and other parameters may be passed to the script using a configuration file or by command line arguments. As a minimum requirement the script requires: i) a list of HiCUP Mapper output file(s) and ii) a digested genome produced by HiCUP Digester.
Configuration File Example:
Digest: Digest_Mouse_Genome_BglII_AluI_11-12-29_08-02-2012.txt
Longest: 800
Shortest: 150
S1_R1_R2_sequence.pair.bam
S2_R1_R2_sequence.pair.bam
S2_R1_R2_sequence.pair.bam
Command Line Example
hicup_filter --digest Digest_Mouse_Genome_BglII_AluI_11-12-29_08-02-2012.txt --longest 800 --shortest 150
The program writes valid Hi-C read pairs to a file named the same as the original, but suffixed with ‘.filt.sam’ or ‘.filt.bam’. Rejected paired sequences are written to different files in a separate date-stamped folder e.g. hicup_filter_ditag_rejects_08-59-17_30-01-2015.
The script also creates a date-stamped file providing an overview of the types of ligation products created e.g. hicup_filter_summary_08-59-17_30-01-2015.txt and further summarises this in SVG-format charts.
Rejected paired reads (Hi-C Experimental Artefacts):¶
A) Sonication protocol:
-Same circularized: DNA fragment cut with the restriction enzyme circularizes, ligating to itself, and is then linearised by sonication
-Same dangling ends: di-tag pairs map to the same restriction fragment and at least one end overlaps the restriction fragment cut site
-Same internal: di-tag maps to a single restriction fragment but neither end of the di-tag overlaps the restriction fragment cut site
-Re-ligation fragments: di-tag pairs map to adjacent restriction fragments which have re-ligated in the same orientation as found in the genome
-Wrong size: calculated di-tag length is not within the limits set by the size-selection step in the experimental protocol
-Contiguous: the di-tag could theoretically represent a contiguous DNA strand spanning several restriction fragments
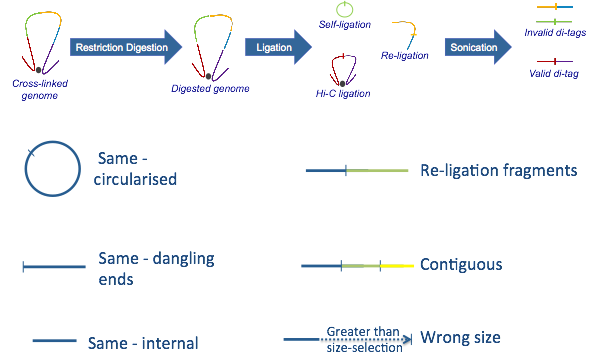
The sonication protocol above is by far the most commonly used procedure, however there is a Hi-C variant where a second restriction enzyme is used to shorten the Hi-C fragments instead of sonicating the samples.
B) Double-digest protocol
-Unmapped: di-tags did not map to expected genomic restriction sites.
-No ligation: read pair members both map to a single restriction enzyme 1 / restriction enzyme 2 double-digest fragment.
-Wrong size: calculated di-tag length is not within the limits set by the size-selection step in the experimental protocol.
-Re-ligation: the original restriction enzyme 1 cut re-anneals. The resulting di-tags map to both the adjacent fragments; one read mapping to one fragment, the other member of the pair mapping to the adjacent fragment.
-Self-ligation: a DNA fragment cut with restriction enzyme 1 circularises, ligating to itself, and is then linearised by the action of restriction enzyme 2.
-Internal restriction enzyme 2 fragments: DNA fragments cut only by restriction enzyme 2 i.e. contain no Hi-C junction or restriction 1 cut sites. Unclassified: while the ends of the fragments map to expected locations within the genome, the orientation of the cut sites do not correspond to any of the aforementioned categories.
Command Line Options¶
--config Specify the optional configuration file --digest Specify the genome digest file (created by hicup_digester) --help Print program help and exit --longest Maximum allowable insert size (bps) --outdir Directory to write output files --quiet Suppress all progress reports --shortest Minimum allowable insert size (bps) --threads Specify the number of threads, allowing simultaneous processing of multiple files --version Print the program version and exit --zip Compress final output files using gzip, or if SAMtools is installed, to BAM format
HiCUP Deduplicator¶
The HiCUP Deduplicator script removes duplicated di-tags (retaining one copy of each) from the data set
Synopsis¶
hicup_deduplicator [OPTIONS]... --config [Configuration FILE]...
hicup_deduplicator [OPTIONS]... [SAM/BAM FILES]...
Function¶
The Hi-C experimental protocol involves a PCR amplification step to generate enough material for sequencing. Consequently, the dataset generated by HiCUP Filter may contain PCR copies of the same di-tag. These PCR duplicates could result in incorrect inferences being drawn regarding the genomic conformation and so should be removed from the data set.
The names of the files to process can be passed to the script either by using a configuration file or command line arguments.
Example:
hicup_deduplicator --zip sample_544_PC_FL_500_lane2.sam
The program creates SAM/BAM files named the same as the input files, only suffixed with ‘.dedup.bam’ or ‘dedup.sam’. If running the whole HiCUP pipeline (not solely the deduplicator script), the final HiCUP file will be end ‘.hicup.bam’ or ‘.hicup.sam’.
De-duplication step only Input: sample_544_PC_FL_500_lane2.filt.sam Output: sample_544_PC_FL_500_lane2.dedup.bam
Input: sample_545_PC_TAM_4_lane3.filt.bam Output: sample_545_PC_TAM_4_lane3.dedup.bam
Whole Pipeline Input: sample_544_PC_FL_500_lane2.filt.bam Output: sample_544_PC_FL_500_lane2.hicup.bam
Input: sample_545_PC_TAM_4_lane3.filt.bam Output: sample_545_PC_TAM_4_lane3.filt.bam
The script also produces a date-stamped summary file (e.g. hicup_deduplicator_summary_08-59-17_30-01-2015.txt) reporting the number of unique di-tags present found in the data set and then classifies those unique di-tags as either cis di-tags (in which both reads are derived from the same chromosome) or trans di-tags (different chromosomes). In addition, this stage of the pipeline produces SVG charts summarising the results.
Command Line Options¶
--config Specify the configuration file --help Print help message and exit --outdir Directory to write output files --quiet Suppress progress reports (except warnings) --threads Specify the number of threads, allowing simultaneous processing of multiple files --version Print the program version and exit --zip Compress output
HiCUP Digester¶
The HiCUP Digester Perl script cuts throughout a selected genome at one or two specified restriction sites
Synopsis¶
hicup_digester [OPTIONS] --config [Configuration FILE]...
hicup_digester [OPTIONS] [FASTA FILES]...
Function¶
The Perl script HiCUP Mapper generates a file of paired mapped reads. While the majority of those reads are expected to be valid Hi-C ligation products, a substantial minority probably will not and should be removed.
The script HiCUP Filter removes many of those invalid pairs, but before it can do this it requires a digested reference genome as input, along with the paired sequence files. The HiCUP Digester program cuts a selected reference genome with one or two specified Type II restriction enzymes that recognise single undivided palindromic sequences. The script prints the results to file for subsequent processing by HiCUP Filter.
The names of the files to be processed and the digestion parameters may be passed to the script by a configuration file or command line arguments. The configuration file contains: i) restriction site 1; ii) restriction site 2 (optional and an atypical choice of protocol); iii) the name of the genome to be processed (optional) and iv) list of FASTA files to be processed.
For example:
re1: A^GATCT,BglII
genome: Mouse
/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37.52.dna.chromosome.1.fa
/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37.52.dna.chromosome.2.fa
/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37.52.dna.chromosome.3.fa
.
.
.
/Genomes/Mouse/NCBIM37/ Mus_musculus.NCBIM37.52.dna.chromosome.Y.fa
The re1 flag refers to the sequence at which the first restriction enzyme used in the Hi-C protocol cuts the genome. The caret symbol (‘^’) marks the position where the enzyme cuts the DNA. As an option the name of the restriction enzyme may be included after the sequence, using a comma to separate the two. Some Hi-C protocols may use two enzymes at this stage to create Hi-C ligation junctions.
Specify two enzymes as follows:
--re1 A^GATCT,BglII:A^AGCTT,HindIII
Restriction site 2 refers to the second, optional (other DNA shearing techniques such as sonication may be used) enzymatic digestion. This restriction site does NOT form a Hi-C ligation junction. This is the restriction enzyme that is used when the Hi-C sonication protocol is not followed. Typically the sonication protocol is followed.
Specify a restriction enzyme to shorten the di-tags instead of sonication (double digest protocol):
--re1 A^GATCT,BglII --re2 AG^CT,AluI
The program creates a tab-delimited file listing:
-column 1: chromosome name (as named in the header row of the FASTA file) -column 2: restriction fragment start position -column 3: restriction fragment end position -column 4: restriction fragment number -column 5: the restriction fragment number if the genome underwent a single digest with restriction enzyme 1 -column 6: the restriction site at the five-prime end of the restriction fragment -column 7: the restriction site at the three-prime end of the restriction fragment
The output file reports the first base for each chromosome as 1 (i.e. NOT 0). The restriction fragment number also starts at 1.
Important note: use the same FASTA files to generate the digested reference genome as to generate the Bowtie indices.
Command Line Options¶
--arima Set the –re1 option to that used by the Arima protocol: ^GATC,DpnII:G^ANTC,Arima --re1 Restriction enzyme used to digest the genome (the enzyme that forms the ligation junction) e.g. A^GATCT,BglII. Some Hi-C protocols may use two enzymes at this stage. To specify two enzymes: -1 A^GATCT,BglII:A^AGCTT,HindIII. --re2 To specify a restriction enzyme instead of sonication to shorten di-tags. This restriction site does NOT form a Hi-C ligation junction. 2 .g. AG^CT,AluI. Typically the sonication protocol is followed. --config Specify the name of the optional configuration file --genome Name of the genome to be digested (not the path to the genome file or files, but the genome name to include in the output file) --help Print program help and exit --outdir Specify the directory to which the output files should be written --quiet Suppress all progress reports --version Print the program version and exit --zip Print the results to a gzip file
HiCUP Reporter¶
The HiCUP Reporter generates summary results for the whole HiCUP pipeline
SYNOPSIS
hicup_reporter [Folder] hicup_reporter [OPTIONS]…
FUNCTION
The hicup_reporter script collates the summary reports generated by the HiCUP pipeline scripts (hicup_truncater, hicup_mapper etc.) and converts them into collated summary text files and interactive documents containing tables and plots.
To run the script, pass as an argument the path to the folder containing the summary files generated by the HiCUP pipeline scripts.
COMMAND LINE OPTIONS
| --help | Print help message and exit |
| --quiet | Suppress progress reports (except warnings) |
| --version | Print the program version and exit |
##References
Processing the Test Dataset¶
To confirm HiCUP functions correctly on your system please download the Test Hi-C dataset. The test files ‘test_dataset1.fastq’ and ‘test_dataset2.fastq’ both contain human Hi-C reads in Sanger FASTQ format.
1) Extract the tar archive before processing:
tar -xvzf hicup_v0.X.Y.tar.gz
2) If necessary, create Bowtie/Bowtie indices of the Homo sapiens GRCh37 genome (chromosomes 1,…,22, X, Y and MT).
Example commands:
bowtie-build 1.fa,2.fa,...,MT.fa human_GRCh37
bowtie2-build 1.fa,2.fa,...,MT.fa human_GRCh37
3) Using HiCUP Digester create a reference genome of Homo sapiens GRCh37 all chromosomes (1,…,22, X, Y and MT) digested with HindIII (A^AGCTT).
Example command:
hicup_digester --genome Human_GRCh37 --re1 A^AGCTT,HindIII *.fa
4) Edit a copy of the hicup.conf configuration file so it has the following parameters:
Zip: 1
Keep: 0
Threads: 1
Bowtie: [Path to Bowtie on your system, if using this aligner]
Bowtie2: Path to Bowtie2 on your system, if using this aligner]
Digest: [Path to digest file on your system]
Index: [Path to Bowtie/Bowtie2 indices on your system]
R: [Path to R on your system]
Format: phred33-quals
Shortest: 50
Longest: 700
test_dataset1.fastq
test_dataset2.fastq
5) Run the pipeline:
Execute HiCUP with the command:
hicup --config [Configuration Filename]
Arima Protocol¶
Arima Genomics has developed a Hi-C kit. HiCUP is compatible with the Arima protocol, simply generate the relevant HiCUP Digest file with the command:
hicup_digester --genome [Genome Name] --arima [FASTA files]
This is all that is additionally required to process an Arima Hi-C dataset.
References¶
Documentation references¶
Lieberman-Aiden et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (326), 289-293
Research that used HiCUP¶
Schoenfelder S, et al. (2018) Divergent wiring of repressive and active chromatin interactions between mouse embryonic and trophoblast lineages. Nat Commun, 9(1):4189
Koohy H, et al. (2018) Genome organization and chromatin analysis identify transcriptional downregulation of insulin-like growth factor signaling as a hallmark of aging in developing B cells. Genome Biol, 5;19(1):126
Aitken SJ, et al. (2018) CTCF maintains regulatory homeostasis of cancer pathways. Genome Biol. 7;19(1):106.
Schoenfelder S, el al. (2018) Promoter Capture Hi-C: High-resolution, Genome-wide Profiling of Promoter Interactions. J Vis Exp. 28;(136).
Montefiori LE, et al. (2018) A promoter interaction map for cardiovascular disease genetics. Elife. 2018 Jul 10;7. pii: e35788.
Choy MK, et al. (2018) Promoter interactome of human embryonic stem cell-derived cardiomyocytes connects GWAS regions to cardiac gene networks. Nat Commun. 28;9(1):2526.
Zhao YT, et al. (2018) Long genes linked to autism spectrum disorders harbor broad enhancer-like chromatin domains. 28(7):933-942
Pan DZ, et al. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity-related genes from GWAS. Nat Commun. 17;9(1):1512.
Baxter JS, et al. (2018) Capture Hi-C identifies putative target genes at 33 breast cancer risk loci. Nat Commun. 9(1), 1028, doi: 10.1038/s41467-018-03411-9
Novo CL, et al. (2018) Long-Range Enhancer Interactions Are Prevalent in Mouse Embryonic Stem Cells and Are Reorganized upon Pluripotent State Transition Cell Rep. 22(10), 2615-2627
Rennie S, et al. (2018) Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat Commun 9(1), 487.
Thomas S, Whalen S, Warburton A, Fernandez SG, McBride AA, Pollard KS, Miranda JJL.
Burren OS, et al. (2017) Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol. 18(1), 165 doi: 10.1186/s13059-017-1285-0
Rubin AJ, et al. (2017) Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet. 49(10), 1522-1528
Petersen R, et al (2017) Platelet function is modified by common sequence variation in megakaryocyte super enhancers. Nat Commun. doi:10.1038/ncomms16058
Nagano T, et al (2017) Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547(7661), 61-67
Siersbaek R, et al. (2017) Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol Cell.66(3):420-435
Freire-Pritchett P, et al. (2017) Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. Elife doi:10.7554/eLife.21926
Moquin SA, et al (2017) The Epstein-Barr virus episome maneuvers between nuclear chromatin compartments during reactivation. Journal of Virology, doi: 10.1128/JVI.01413-17
Sud A, et al. (2017) Genome-wide association study of classical Hodgkin lymphoma identifies key regulators of disease susceptibility. Nature Communications 8(1), 1892
Ke Y et al. (2017) 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 170(2):367-381
Litchfield K, et al. (2017) Identification of 19 new risk loci and potential regulatory mechanisms influencing susceptibility to testicular germ cell tumor. Nature Genetics 49(7), 1133-1140
Gabriele M et al, (2017) YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. American Journal of Human Genetics 100(6), 907-925
El-Sharnouby S et al. (2017) Regions of very low H3K27me3 partition the Drosophila genome into topological domains. PloS one 12(3), e0172725
Law PJ, et al. (2017) Genome-wide association analysis of chronic lymphocytic leukaemia, Hodgkin lymphoma and multiple myeloma identifies pleiotropic risk loci. Scientific Reports 7(41071)
Aymard F, et al. (2017) Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nature Structural & Molecular Biology 24(4), 353-361
Javierre BM, et al. (2016) Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters Cell 167(5), 1369-1384
Cairns J, et al. (2016) CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 17(1), 127
Andrey G, et al. (2016) Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Research 27(2), 223-233
Franke M, et al. (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538(7624), 265-269
Krueger F, et al. (2016) SNPsplit: Allele-specific splitting of alignments between genomes with known SNP genotypes. F1000Research 5:1479 DOI: 10.12688/f1000research.9037.2
Wu HJ, et al. (2016) A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 32(24), 3695-3701
Veluchamy A, et al. (2016) LHP1 Regulates H3K27me3 Spreading and Shapes the Three-Dimensional Conformation of the Arabidopsis Genome. PLoS One 11(7):e0158936
Schoenfelder S, et al. (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nature Genetics, 47(10), 1179–1186
Mifsud B, et al. (2015) Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics, 47(6), 598-606
Nagano T, et al. (2015) Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biology, 16(1), 175
Sahlén P, et al. (2015) Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biology, 16(1), 156
Schoenfelder, S. et al. (2015) The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research, 25(4), 582-97
Chandra, T. et al. (2015) Global Reorganization of the Nuclear Landscape in Article Global Reorganization of the Nuclear Landscape in Senescent Cells. Cell Reports, 10(4), 1–13
Dryden, N. H. et al. (2014) Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Research, 24(11), 1854-1868
Acknowledgements¶
HiCUP was written by Steven Wingett (Bioinformatics Group, Babraham Institue, Cambridge, UK).
Rola Dali, Edouard Henrion and Mathieu Bourgey (McGill University, Canada) assisted with making HiCUP compatible with the Arima protocol.
Terms of use¶
HiCUP is distributed under a “GNU General Public License”, a copy of which is distributed with the software.
Report problems¶
Please email steven.wingett@babraham.ac.uk if you have any problems running this program.
How to cite HiCUP¶
HiCUP is described in:
Wingett S, et al. (2015) HiCUP: pipeline for mapping and processing Hi-C data F1000Research, 4:1310 (doi: 10.12688/f1000research.7334.1)