
FastQ Screen
| Function | FastQ Screen allows you to screen a library of sequences in FastQ format against a set of sequence databases so you can see if the composition of the library matches with what you expect. |
|---|---|
| Language | Perl |
| Requirements | Linux-based operating system Bowtie or Bowtie2 or BWA gzip (optional) SAMtools (optional) GD::Graph (optional) Bismark (bisulfite mapping only) |
| Code Maturity | Stable - has been working in production for some time |
| Code Released | Yes, under GPL v3 or later. |
| Documention | Online documentation here |
| Online Tutorials | Online tutorials here |
| Initial Contact | Steven Wingett |
Download Now |
|
![]()
When running a sequencing pipeline it is useful to know that your sequencing runs contain the types of sequence they're supposed to.
FastQ Screen allows you to set up a standard set of libraries against which all of your sequences can be searched. Your search libraries might contain the genomes of all of the organisms you work on, along with PhiX, Vectors or other contaminants commonly seen in sequencing experiments.
Click here for a video introduction to FastQ Screen.
The program produces both text based and graphical output which summaries the mapping of your sequences against each of your libraries, so that when you search your mouse sequences you can see if they're good, like this:
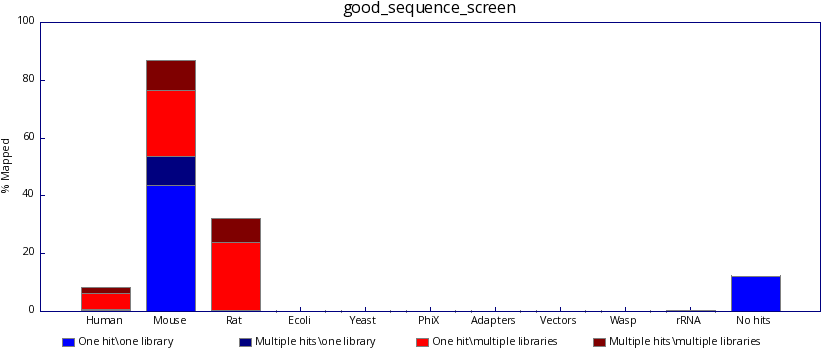
..and not bad, like this:
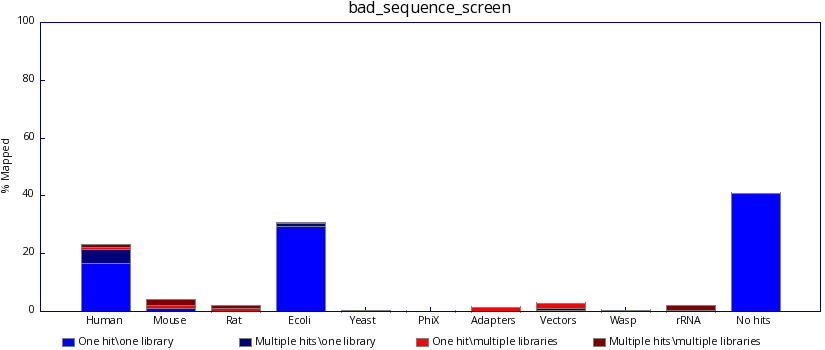
Changelog
- Click here for notes on all releases Version 0.14.1 onwards
- 17-06-19: Version 0.14.0 released
-
- New --add_genome option
- Fixed bug causing FastQ Screen to run when aligner not present
- 12-09-18: Version 0.13.0 released
-
- Added --get_genomes option
- Updated documentation
- 16-08-18: Version 0.12.2 released
-
- For bisulfite mapping, Bismark is now run in --ambiguous mode to identify multi-mapping reads
- 18-06-18: Version 0.12.1 released
-
- Added --inverse option for filtering files
- Improved results display format
- 18-06-18: Version 0.12.0 released
-
- Interactive HTML graphs now made using Plotly
- 27-11-17: Version 0.11.4 released
-
- FASTQ files are edited so that the third line of a read is always a plus symbol, therby preventing tagged/filtered output files not technically adhering to FASTQ format
- 16-10-17: Version 0.11.3 released
-
- FastQ Screen uses full path to dependencies rather than Bowtie, Bowtie2 etc.
- 21-09-17: Version 0.11.2 released
-
- Fixed bug preventing --tag being selected without --filterng selected without --filter.Fixed bug preventing --tag being selected without --filter
- In bisulfite mode, FastQ Screen no longer assumes that Bowtie/Bowtie2 are always in the path (even if specified otherwise in the config file)
- FastQ Screen now terminates before creating a subset file if no aligner/Bismark executable file is found
- FastQ Screen no longer gives an initialisation warning if, in the configuration file, the DATABASE line does not specify a database name and/or database path
- 23-02-17: Version 0.11.1 released
-
- Fixed bug preventing selection of --filter options 4 or 5
- 22-02-17: Version 0.11.0 released
-
- Added --filter options 4 and 5
- Added option --pass to further improve filtering
- Prevented HTML bar graphs overlapping when screening against multiple genomes
- Corrected documentation describing how to use the option --top
- 19-01-17: Version 0.10.0 released
-
- Added option --top for faster processing, when speed of processing is the highest priority
- Improve appearance of HTML graphs
- 07-12-16: Version 0.9.5 released
-
- Fixed bug causing FastQ Screen, when running in --bisulfite mode, to mislabel species names in the HTML bisulfite read orientation graph on the occasion that one or more of the samples tested contained reads that mapped to none of the bisulfite converted reference genomes
- 21-11-16: Version 0.9.4 released
-
- New colour scheme which should be easily interpretable by colour blind people
- Fixed bug preventing, in some instances, genome names being displayed in the header line of filtered (using the --filter option) FASTQ files
- 31-10-16: Version 0.9.3 released
-
- Fixed bug stopping the command-line --threads option overriding the configuration file
- When the --tag option was specified, FastQ Screen should have analysed all the reads in a file by default. However, a bug resulted in a subset file being generated and analysed instead. This has been fixed and now a reduced reads file will only be generated with the --tag option if explicitly requested using the --subset command
- Fixed bug causing FastQ Screen to not process reads containing a full-stop in the FASTQ read header
- Changes to what is reported when using the --quiet option
- Updated documentation
- 12-10-16: Version 0.9.2 released
-
- When --outdir option selected, FastQ Screen creates the output directory if it does not already exist
- FastQ Screen in bisulfite mode checks whether the bisulfite orientation graph already exists
- Adjusted how compressed files are read to improve compatibility with Mac systems
- Fixed bug causing the HTML report to be missing one genome when reporting conventional (i.e. not bisulfite) alignment results
- Updated documentation
- 05-10-16: Version 0.9.1 released
-
- Fixed bug causing FastQ Screen to terminate prematurely when in bisulfite mode if no reads map to a bisulfite reference genome
- 04-10-16: Version 0.9.0 released
-
- FastQ Screen, when run in Bisulfite mode, reports to which strand reads aligned (original top strand, complementary to original top strand, complementary to original bottom strand, or original bottom strand)
- 08-09-16: Version 0.8.0 released
-
- Program is now compatible with aligner BWA
- FastQ Screen produces an HTML summary report
- Program documentation has been substantially updated and is now in Markdown format
- 01-08-16: Version 0.7.0 released
-
- Added --tag option to create output FASTQ files in which the the genomes to which a read maps is appended to the first line of the FASTQ read
- Added --filter option to extract reads from a tagged FASTQ file which map, or do not map, to a specified combination of genomes
- Pre-existing option --nohits is now equivalent to the parameters --tag --filter 000 (number of zeroes corresponds to the number of genome being screened)
- 12-07-16: Version 0.6.4 released
-
- Program no longer terminates if a single Bismark reference genome index is incorrectly specified
- Fixed bug causing program to crash if --aligner bowtie2 and --bisulfite specified together
- FastQ Screen can now use Bowtie (in addition to Bowtie2) when performing Bisulfite mapping with Bismark
- Fixed bug in how FastQ Screen checks for dependencies (e.g. SamTools)
- 07-07-16: Version 0.6.3 released
-
- Fixed bug causing --subset 0 to crash
- Fixed bug in which the reported percentage reads mapping to no libraries was, in some instances, an underestimate of the correct value
- 05-07-16: Version 0.6.2 released
-
- Updated help text
- Refactored code
- 01-07-16: Version 0.6.1 released
-
- Fixed bug causing program to crash in some instances when --outdir option selected
- 27-06-16: Version 0.6.0 released
-
- Compatible with Bismark, enabling bisulfite library QC
- Option --colorspace is no longer supported
- 07-09-15: Version 0.5.2 released
-
- Fixed bug observed when --nohits option selected causing initialization warnings, in some instances
- 14-07-15: Version 0.5.1 released
-
- Ensures a FASTQ file is not mapped against the same library more than once
- 29-06-15: Version 0.5.0 released
-
- Please note that users no longer need to specify whether a genome index is compatible with bowtie or bowtie2, since this is now determined automatically
- Option --subset 100000 is now the default. Use --subset 0 to process an entire file (not recommended for most QC applications, since this generally takes much more time)
- Option --paired removed
- Bowtie2 is now the default aligner, replacing the orignal bowtie
- New option --force instructs fastq_screen to overwrite extant output files
- The script now uses a more memory efficient internal data structure for recording which reads map to what library. However, this means that a maximum of 15 libraries may be specified with 32-bit Perl or 31 libraries with 64-bit Perl
- 09-07-14: Version 0.4.4 released
-
- Fixed an improper check for bowtie2 indices for large genomes
- Added a check for an even number of input files if --paired is specified
- Improved output file name generation for .fq and .gz files
- Improved the appearance of output graphs
- 03-06-14: Version 0.4.3 released
-
- Fixed bug causing all reads to be written to the 'no hits' output file when using Bowtie2 as the aligner
- The 'nohits' output file has the file extension '.fastq' and is compressed if the input files are compressed
- 30-09-13: Version 0.4.2 released
-
- The script no longer defaults to Bowtie if '--aligner' is not specified but instead intelligently selects an appropriate aligner
- Reports the number of reads mapping each genome in addition to percentages
- 13-06-13: Version 0.4.1 released
-
- Command line argument (--aligner, -a) introduced, enabling the user to select the sequence aligner
- Skips the production of graphs if GD::Graph is not installed
- Standard error now reported as a separate file for each library
- 12-12-12: Version 0.4 released
-
- FastQ Screen now compatible with Bowtie2
- 18-4-12: Version 0.3.1 released
-
- Fixed bug preventing use of the Bowtie argument (--bowtie, -b)
- 29-3-12: Version 0.3 released
-
- The --multilib option was removed. This script now returns simultaneously all the information previously obtained by selecting/not selecting --multilib
- Added --nohits option which prints to an output file sequence reads or read pairs that mapped to none of the reference genomes
- Option --illumina flag has changed to --illumina1_3
- Script can process files compressed with gzip
- 19-5-11: Version 0.2.1 released
-
- Fixed a bug in multilib paired end searches which caused reported mapped percentages to be double the true value
- 18-5-11: Version 0.2 released
-
- Added --multilib option for better comparisons between genomes
- Added colorspace support
- Allow override of default config file
- 24-03-11: Version 0.1 released